
Researchers Study The Role of Dystrophin Related Proteins in Progression of Muscular Dystrophy Disease
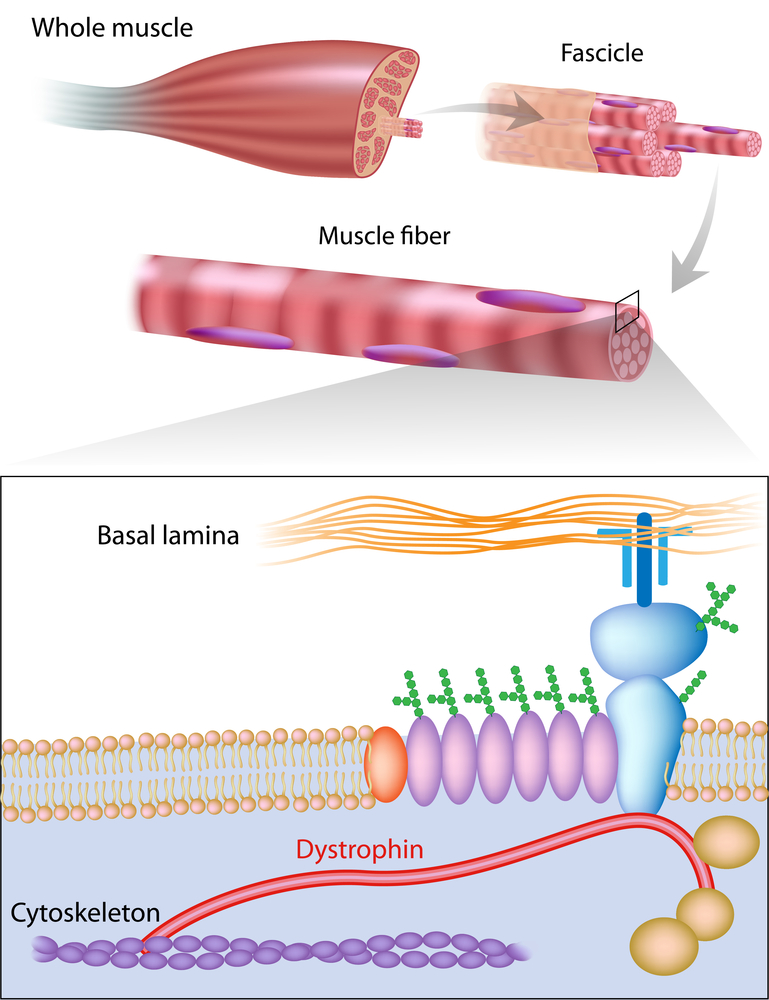



In a recent paper published in Scientific Reports entitled “Deletion of integrin α7 subunit does not aggravate the phenotype of laminin α2 chain-deficient mice“, researchers from Sweden have revealed novel insights into the role of dystrophin related proteins towards the progression of muscular dystrophy (MD).
Muscular dystrophy is a disease characterized by weakened musculoskeletal system. Generally, the process occurs progressively through the weakening of skeletal muscle, inducing deficiency in muscle proteins that later leads to the death of muscle cells and tissue. Patients with MD suffer from various symptoms like curvature of the spine and the back, frequent falls, inability to walk, muscle spasms, progressive muscular wasting, poor balance, drooping eyelids, joint contractures, and respiratory difficulties. MD comes in various forms with the nine most prominent types accounting for Becker, limb-girdle, congenital, facioscapulohumeral, myotonic, oculopharyngeal, distal, and Emery-Dreifuss MD. The difference between these types of MD lies in the specific body muscle that is affected. For example, while Emery–Dreifuss MD affects skeletal and heart muscles, distal MD affects hand or feet muscles.
It is generally assumed that MD is genetically inherited. Nevertheless, 33% of MD cases are not related to generic inheritance, rather to impairment or mutations in the dystrophin protein or associated complexes. Biochemically, dystrophin is an important part of a protein complex which links the cytoskeleton of a muscle fiber to the surrounding extracellular matrix through the cell membrane. Among these molecules, laminin-211 is a protein of the extracellular matrix composed of subunits α2, β1 and γ1 that biologically binds to a part of the dystrophin protein complex named integrin α7β1 receptor.
It is believed that these proteins play key roles in muscle function and their respective deficiency yield different forms of muscular dystrophies with variable severity in humans and animal models. In this study the research team analyzed if the laminin-211 protein and the integrin α7 receptor played vital roles in normal muscular function. As such, they deleted both molecules in mice models, showing that absence of the integrin α7 receptor did not aggravate the features of mice deficient for laminin α2. In other words, the mice had similar survival rate, weight, and MD biopsy with equal inflammation and fibrosis.
RELATED: Engineered Muscle May Fix Muscle Wasting in Muscular Dystrophy
In conclusion, contrary to what is believed (that integrin subunit α7 may have a significant role in modifying disease progression in muscular dystrophies), these findings demonstrate that integrin α7β1 plays no role in skeletal muscle other than to mediate laminin α2 chain interactions. Furthermore, it is possible that laminin α2 chain may interact with other receptors, a theory which the team plans to explore in future experiments. This information may pave the way to further understand the causes/mechanisms of MD disease progression.


