


Duchenne Muscular Dystrophy: Clinical Presentation and Key Differentiators
Written by Margaret Anne Rockwood | Last updated June 4, 2026
 Medically reviewed by Daniel Guillen, MD
Medically reviewed by Daniel Guillen, MD
Duchenne Muscular Dystrophy: Clinical Presentation and Key Differentiators
Duchenne muscular dystrophy (DMD) is the most common and severe form of childhood-onset muscular dystrophy. Early recognition and differentiation from other muscular dystrophies and neuromuscular mimics are critical for timely genetic confirmation, multidisciplinary management, and initiation of therapy. Given the expanding therapeutic landscape—including exon-skipping agents and gene therapies—early diagnosis has become increasingly important.
Clinical Presentation
Early Developmental Signs
Symptoms typically manifest between ages 2 and 5 years. Early indicators are often subtle and may include:
- Delayed motor milestones (for example, walking after 18 months)
- Difficulty running, jumping, or climbing stairs
- Frequent falls
- Toe walking
Some children initially present with isolated speech delay, behavioral concerns or elevated transaminases before overt weakness becomes clinically apparent,
Progressive Proximal Weakness
The hallmark of DMD is symmetric proximal muscle weakness, particularly affecting:
- Hip extensors and abductors
- Quadriceps
- Shoulder girdle muscles (later in the disease course)
- Reduced endurance or “slower” than peers
A classic clinical sign is the Gowers maneuver, in which the child uses their hands to “climb up” their thighs to stand from the ground. Although, this may not be as obvious in earlier stages of the disease.
Red Flags Suggesting Duchenne Muscular Dystrophy
Physicians should maintain a high index of suspicion when encountering:
- Male child with delayed motor milestones
- Gowers’ sign
- Calf pseudohypertrophy
- Family history of early male deaths or wheelchair use
- Persistent elevated CK without clear cause
Early referral for genetic testing is recommended when these features are present.
Calf Pseudohypertrophy
Enlargement of the gastrocnemius muscles is a characteristic feature. Pseudohypertrophy reflects fatty and fibrotic replacement of muscle tissue rather than true muscle hypertrophy.
Loss of Ambulation
Most untreated patients lose independent ambulation by ages 10–12 years. Contractures, scoliosis, and progressive weakness contribute to declining mobility. In contrast, corticosteroid-treated patients receiving modern multidisciplinary care may maintain ambulation substantially longer.
Cardiopulmonary Involvement
- Cardiomyopathy: Dilated cardiomyopathy is common and may be asymptomatic .
- Respiratory decline: Nocturnal hypoventilation may initially present subtly with morning headaches, daytime somnolence, fatigue or impaired concentration. Progressive diaphragmatic weakness leads to restrictive lung disease and eventual ventilatory insufficiency.
Laboratory and Diagnostic Findings
- Markedly elevated serum creatine kinase (CK), often 10–100 times normal
- Genetic testing confirming a dystrophin gene mutation
- Muscle biopsy (rarely needed today) demonstrating absence of dystrophin
- Elevated transaminases, often misdiagnosed as liver disease, may be an incidental finding
Differentiating Duchenne from Other Muscular Dystrophies
Common non-Duchenne muscular dystrophies include Becker muscular dystrophy (BMD), limb-girdle muscular dystrophies (LGMD), facioscapulohumeral muscular dystrophy (FSHD), myotonic dystrophy, congenital muscular dystrophies, and Emery–Dreifuss muscular dystrophy.
Accurate differentiation relies on age of onset, severity, pattern of weakness, and progression rate.
1. Duchenne vs. Becker Muscular Dystrophy
Scroll horizontally to view all columns -->
| Feature | Duchenne MD | Becker MD |
| Onset | Early childhood (2–5 years) | Adolescence or adulthood |
| Dystrophin | Absent | Reduced or abnormal |
| Progression | Rapid | Slower |
| Loss of ambulation | Around 10–12 years | Often into adulthood |
| Cardiomyopathy | Common, often early | Common, may dominate presentation |
High-level differentiator: If a patient remains ambulatory into late adolescence with a similar distribution of weakness, Becker muscular dystrophy is more likely.
Of note, many in the field have begun to consider DMD and BMD as dystrophinopathies that represent a spectrum of disease. This is partially because patients with genetic diagnoses of either DMD or BMD can have milder or more severe courses than what would be expected.
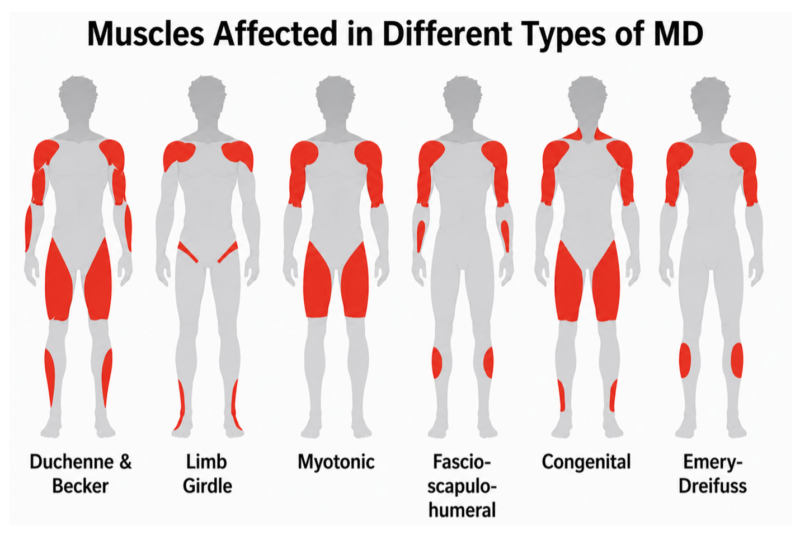
2. Duchenne vs. Limb-Girdle Muscular Dystrophies
Limb-girdle muscular dystrophy (LGMD) comprises a heterogeneous group of disorders affecting predominantly proximal muscles.
Key Differences:
- Inheritance: LGMD may be autosomal dominant or autosomal recessive; DMD is X-linked.
- Onset: LGMD varies widely, from childhood to adulthood.
- Distribution: Both cause proximal weakness, but LGMD often lacks early calf pseudohypertrophy.
- CK levels: Elevated in both, but often less extreme in LGMD.
- Cognitive involvement: Typically absent in LGMD.
High-level differentiator: A family history involving both sexes or an autosomal inheritance pattern should prompt consideration of LGMD over DMD.
3. Duchenne vs. Myotonic Dystrophy
Myotonic dystrophy differs fundamentally from Duchenne muscular dystrophy in both the pattern of weakness and systemic involvement.
Key Differences:
- Muscle involvement: Myotonic dystrophy typically presents with distal muscle weakness involving the hands, forearms, and lower legs. In DMD, predominantly affects proximal muscles, especially the pelvic girdle. In children with both diseases, hypotonia, developmental delay and facial weakness are common early symptoms.
- Myotonia: A hallmark of myotonic dystrophy is myotonia (delayed muscle relaxation), often evident as grip stiffness or difficulty releasing a handshake. This is not present in DMD.
- Onset: Myotonic dystrophy usually presents in adolescence or adulthood (although congenital forms exist) later, whereas DMD presents in early childhood.
- Systemic involvement: Myotonic dystrophy is a multisystem disorder commonly associated with cardiac conduction abnormalities, cataracts, endocrine dysfunction, and cognitive changes. DMD primarily affects skeletal and cardiac muscle, with some neurodevelopment involvement.
- Progression: Myotonic dystrophy often follows a slower, more variable course, while DMD is rapidly progressive with early loss of ambulation.
4. Duchenne vs. Facioscapulohumeral Muscular Dystrophy
Facioscapulohumeral muscular dystrophy (FSHD) presents with a distinct pattern of weakness.
Key Differences:
- Muscle involvement: FSHD prominently affects facial muscles (for example, inability to whistle or incomplete eye closure) though this may initially be subtle. Scapular winging may precede overt proximal arm weakness.
- Onset: Often adolescence or early adulthood.
- Progression: Slower and more variable.
- Lower limb involvement: Occurs later, unlike the early lower-limb weakness seen in DMD, often causing foot drop, which is not a common feature of DMD.
High-level differentiator: Facial weakness is not a feature of DMD; its presence should strongly suggest FSHD,
5. Duchenne vs. Congenital Muscular Dystrophies
Congenital muscular dystrophies present at or shortly after birth.
Key Differences:
- Onset: Neonatal hypotonia (“floppy infant”) in congenital forms versus delayed onset in DMD.
- Motor milestones: Often never fully achieved in congenital muscular dystrophies.
- Brain involvement: Structural brain abnormalities are more common.
High-level differentiator: Normal infancy followed by delayed milestones and progressive weakness favors DMD over congenital forms.
6. Duchenne vs. Emery–Dreifuss Muscular Dystrophy
Emery–Dreifuss muscular dystrophy has a distinct humeroperoneal distribution and characteristic early contractures that distinguish it from DMD.
Key Differences:
- Muscle involvement: Emery–Dreifuss typically affects humeral (upper arm) and peroneal (lower leg) muscles, whereas DMD primarily involves proximal muscles, especially the hips and thighs.
- Contractures: Early joint contractures involving the elbows, Achilles tendons, and posterior neck are a hallmark of Emery–Dreifuss and may precede significant weakness. In DMD, contractures occur later.
- Onset: Emery–Dreifuss often presents in late childhood to adolescence, compared with early childhood onset in DMD.
- Cardiac involvement: Emery–Dreifuss is strongly associated with cardiac conduction defects such as arrhythmias and heart block, often preceding and manifesting with more severity than skeletal muscle weakness. DMD more commonly causes dilated cardiomyopathy.
- Progression: Emery–Dreifuss generally progresses more slowly, with preservation of ambulation into adulthood, whereas DMD is rapidly progressive.
High-level differentiator: Early elbow or Achilles contractures with humeroperoneal weakness should prompt consideration of Emery–Dreifuss muscular dystrophy rather than Duchenne.
Why Different Muscular Dystrophies Affect Different Muscles
Although often grouped together, muscular dystrophies do not share a single mechanism. Only dystrophinopathies—such as Duchenne muscular dystrophy and Becker muscular dystrophy—result from loss of dystrophin. Other forms involve distinct proteins and cellular pathways, leading to selective patterns of muscle involvement.
Several factors explain this regional vulnerability:
- Protein function and localization: Different muscular dystrophies affect proteins with specialized roles such as membrane stability, nuclear structure, or RNA processing. These functions are not equally critical across all muscle groups.
- Mechanical stress: Muscles subjected to greater biomechanical load are more susceptible to injury when structural integrity is compromised. This helps explain early proximal weakness in dystrophinopathies and contracture-prone regions in disorders such as Emery–Dreifuss muscular dystrophy.
- Muscle fiber composition: Variations in type I (slow-twitch) and type II (fast-twitch) fiber distribution influence susceptibility, as certain diseases preferentially affect specific fiber types.
- Regenerative capacity: Muscle groups differ in their ability to repair damage. Limited regenerative reserve may contribute to early involvement of specific regions, such as facial muscles in FSHD.
- Gene expression patterns: Disease-related genes may be differentially expressed across muscle groups, contributing to characteristic distributions of weakness.
Clinical Implication
Recognizing the pattern of muscle involvement is central to diagnosis. Distinct distributions—proximal, distal, facial-scapular, or humeroperoneal—reflect the underlying biology and can guide targeted evaluation.
Pattern Recognition Keys
-
- Proximal weakness → think DMD/LGMD
- Facial + scapular → think FSHD
- Distal + myotonia → think myotonic
- Humeroperoneal + contractures → think EDMD
Summary Evaluation Steps
- Serum CK measurement (initial screening)
- Genetic testing (multiplex ligation-dependent probe amplification or sequencing)
- Cardiac evaluation (baseline echocardiogram or cardiac MRI)
- Pulmonary function testing (age-appropriate)
Muscle biopsy is now generally reserved for atypical or inconclusive cases. Increasingly, diagnosis occurs through family screening, incidental hyperCKemia, or newborn screening initiatives are gaining momentum.
References
- Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Birnkrant, D. J., et al. (2018). The Lancet Neurology, 17(4), 347–361.
- DMD gene and dystrophinopathy phenotypes associated with genetic testing results. Andrews, J. G., et al. (2023). Applied Clinical Genetics, 16, 81–98.
- Diagnosis and management of Becker muscular dystrophy. Magot, A., et al. (2023). Current Opinion in Neurology, 36(5), 574–581.
- Dystrophinopathies. Darras, B. T., Miller, D. T., & Urion, D. K. (1993/updated). In GeneReviews®.
- Hereditary muscular dystrophies and the heart. Hermans, M. C. E., et al. (2010). Neuromuscular Disorders, 20(8), 479–492.
- Myotonic dystrophy type 1 or Steinert’s disease. Romeo, V. (2012). Advances in Experimental Medicine and Biology, 724, 239–257.
- Facioscapulohumeral muscular dystrophies. Wagner, K. R. (2019). Continuum (Minneap Minn), 25(6), 1662–1681.
- Facioscapulohumeral muscular dystrophy. Mul, K., et al. (2022). Current Opinion in Neurology, 35(5), 614–620.
- Clinical aspects of Emery-Dreifuss muscular dystrophy. Madej-Pilarczyk, A. (2018). Nucleus, 9(1), 268–274.
- Genetic basis of limb-girdle muscular dystrophies: The 2014 update. Nigro, V., & Savarese, M. (2014). Acta Myologica, 33(1), 1–12.
- Consensus-based care recommendations for adults with myotonic dystrophy type 1. Ashizawa, T., et al. (2018). Continuum/Neurology.
- Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Ricotti, V., et al. (2016). Developmental Medicine & Child Neurology, 58(1), 77–84.